

基因敲除、基因敲低、基因敲入都是常见的基因功能研究方法,虽然名称很相似,但其实验方法和效果却截然不同今天,我们就结合图文一起来了解一下这三种技术吧!
一、基因敲除
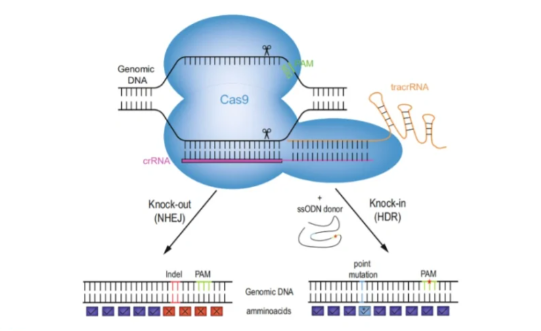
原理
是用含有一定已知序列的小DNA片段与受体细胞基因组中序列相同或相近的基因发生同源重组,整合至受体细胞基因组中并得到表达的一种外源DNA导入技术。
载体
①CRISPR/Cas9载体:通过Cas9核酸酶和SgRNA靶向序列精确定位并靶向基因DNA序列,然后引入双链断裂点,完成基因敲除。
②TALENS载体:包含特异性核酸酶和nuclease binding domain,通过切割靶向基因DNA序列,完成敲除和编辑。
③ZFNS载体:包含具有特异性的锌指结构域和核酸酶,通过切割靶向基因DNA序列,完成敲除和编辑。
载体构建方法
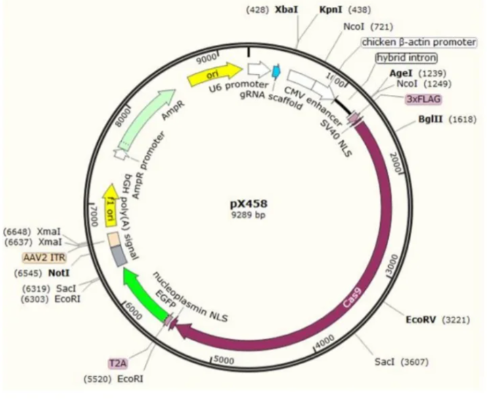
①设计和选择:设计基因敲除的靶点,选择CRISPR/Cas9等工具进行编辑。
②载体插入:将编辑过的靶点序列插入到特定的载体中。
③转化和验证:转化编辑好的敲除载体至大肠杆菌中,进行筛选鉴定,验证敲除效果。
④导入目标细胞:通过病毒载体、电穿孔等方法将敲除载体导入到目标细胞中。
二、基因敲低
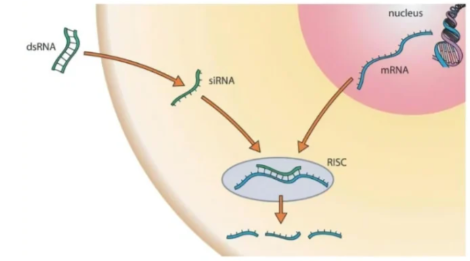
原理
基因敲低又称基因敲降,是利用抑制剂来抑制基因的表达过程。有几种不同的抑制方式可用于抑制基因的表达,其中主要的抑制方式有RNA干扰技术(RNAi),CRISPR-Gas9技术以及基因转染技术。这些技术使用不同的原理,但它们的共同点是都能够发挥基因敲低的效果。
载体
①SiRNA载体:短干扰RNA。在3'端有两个碱基的游离,可激活RNA干扰,通过与目标mRNA互补结合序列,特异性地实现靶向mRNA降解。
②ShRNA载体:短发夹RNA。一种SiRNA前体,包括一个“茎”和一个“环”结构,由RNA序列的短区域(这些区域形成“茎”)之间的碱基配对形成,由不形成碱基对的短序列(形成“环”)隔开。
③CRISPRi载体:包含CRISPRi靶向序列和转录抑制子,通过抑制靶向基因的转录水平实现基因敲低效果。
载体构建方法
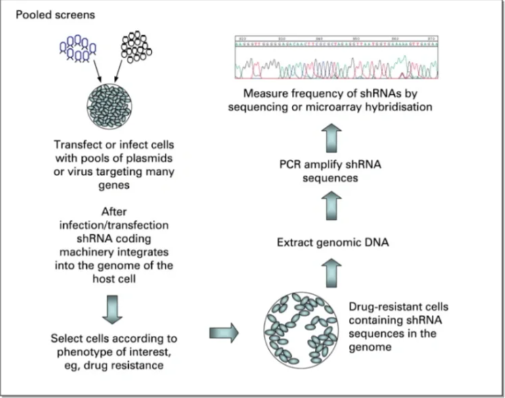
①设计干扰序列:采用在线设计工具或经验公式来计算。
②合成和克隆:合成干扰序列的DNA或RNA小片段,并克隆到转录载体中,生成SiRNA或shRNA表达载体。
③转化和筛选:将siRNA或shRNA表达载体转化至大肠杆菌中,进行筛选鉴定。
④导入目标细胞:通过病毒等途径将重组质粒导入目标细胞中。
三、基因敲入
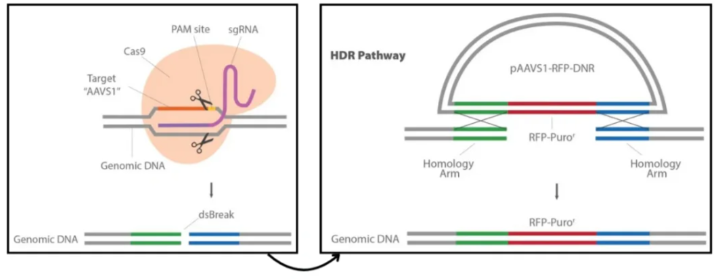
原理
利用基因同源重组,将外源的功能基因(基因组原先不存在、或已失活的基因)转入细胞与基因组中的同源序列进行同源重组,插入到基因组中,在细胞内获得表达的技术。
载体
①SgRNA-Cas9载体:在KI基因编辑系统中像一把“剪刀”,可以精准切开双链DNA,这把“剪刀”锋利与否和精确度能直接影响KI基因编辑系统的编辑效率。
②Donor DNA:一段高度同源的DNA模板。以Donor DNA为模板进行修复,可以将目的片段定点引入基因组。
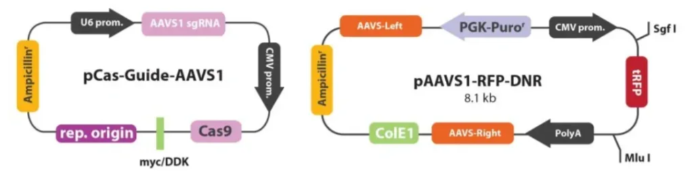
载体构建方法
①设计sgRNA及修复DNA模版:为了保证sgRNA没有预测的脱靶结合位点,根据人类AAVS1 AAVS1 Safe Harbor基因设计SgRNA,并进行软件分析。设计DNA修复模版,其两侧各有600bp的同源臂。
②)构建sgRNA质粒及修复DNA模板质粒:选定sgRNA设计与CMV启动子驱动的Cas9基因,将其克隆到pCas-Guide中来制备pCas-Guide-AAVS1;再将DNA修复模板插入含有RFP-嘌呤霉素的pAAVS1-RFP-DNR表达载体。
③将sgRNA-Cas9、修复DNA模版共转293T细胞:用lipofectmine2000转染试剂将上述两种质粒转染至293T细胞。
 文章推荐
文章推荐

业务咨询
业务咨询专线:133 7682 0615
Email:lxyjy@wie-biotech.com


