

大家做qRT-PCR检测时,可能会经常看到一些非常奇怪的扩增图,并感到一头雾水。下面小编为大家整理了qRT-PCR中常见的10个问题及其解决方法。
一、如何判断qRT-PCR数据有效性
扩增曲线
应展现出一条平滑的S型扩增曲线,起始阶段无扩增现象,起峰时间符合顺利达到扩增平台期的正常标准。阳性结果的Ct值应调整在15到30之间,而阴性对照Ct值应大于35,内参基因的Ct值小于20,不同样品之间内参基因的Ct值差异不超过1个单位,对于复孔Ct值标准偏差在0.5以内。
溶解曲线
应呈现宽度较小的单峰,表明是特异性扩增
标准曲线
斜率范围在-3.58至-3.10之间。引物扩增效率保持在90%至110%之间,这意味着引物的扩增能力相对稳定,并且效率较高。R2值通常用来表示数据点与回归线之间的拟合程度,当R2值大于0.98时,意味着实验数据的线性关系非常强。
二、扩增曲线不稳定

可能原因:
1. RNA纯度不足,未达到实验所需标准。
2. 体系中杂质含量过高,影响实验效果。
3. 仪器长时间使用,性能可能出现偏差。
解决方法:
1. 遵循实验流程,确保提取的RNA纯度达到实验要求。
2. 对体系中的杂质进行严格的控制和除去。
3. 定期对仪器进行校准和维护,确保仪器性能稳定。
三、扩增无法达到平台期
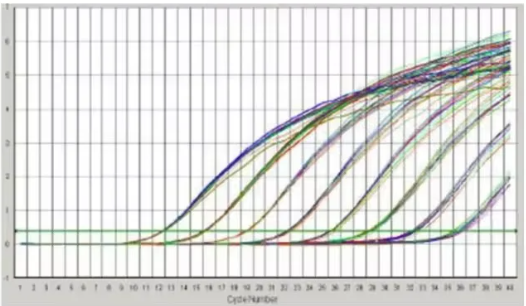
可能原因:模板丰度低;循环数过少;试剂扩增效率不佳。
解决方法:
1. 为确保实验准确性,建议适当提升模板的添加量,以增加模板浓度。
2. 适当增加循环次数,以确保充分扩增。
3. 通过标准曲线测定方法,适当调整镁离子浓度,并考虑更换为扩增效率更高的试剂。
四、曲线下降
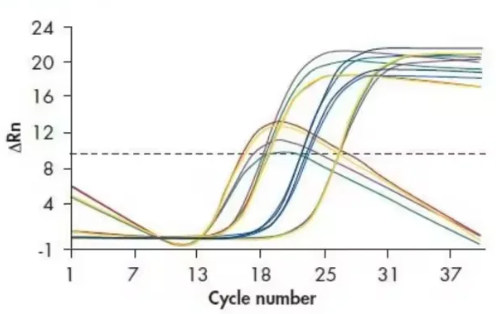
可能原因:
1. 体系中存在降解现象;
2. 模板浓度超过了适宜范围,导致浓度过高。
解决方法:
1. 提高体系纯度,以减少降解的发生;
2. 适当减少模板量,使其达到合适的浓度范围;
3. 在确保实验准确性的前提下,适当降低荧光阈值,以优化实验结果。
五、单峰但不尖锐
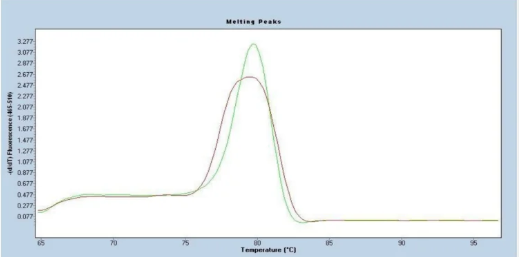
可能原因:
1. 试剂成分的特异性可能影响实验结果。
2. 存在与目标序列大小相近的非特异性扩增产物。
解决方法:
1. 若实验过程中的温度跨度未超过7℃℃,则可将实验结果视为有效。
2. 建议进行高浓度琼脂糖电泳,以确认是否形成单一条带,进而验证实验结果的准确性。
六、单峰,Tm低于80℃
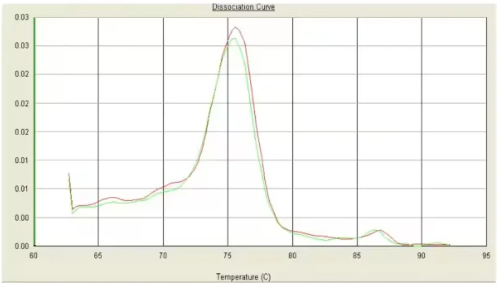
可能原因:
1. 存在引物二聚体。
2. 扩增的片段长度过短。
解决方法:
1. 首先确定实验过程中是否已正确加入模板,以确保实验条件符合预设要求。
2. 建议进行琼脂糖电泳检测,以验证扩增产物条带的大小是否符合预期。
七、双峰,较低峰Tm在80℃之前
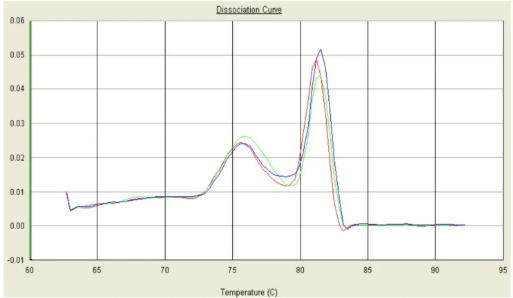
可能原因:模板浓度不足或引物浓度偏高可能导致引物之间非特异性结合,进而形成引物二聚体。
解决方法:
1. 适当提高退火温度,以减少非特异性结合发生。
2. 增加模板的投入量,降低引物浓度,优化反应体系。
3. 重新设计引物。
八、双峰,Tm都在80℃之后
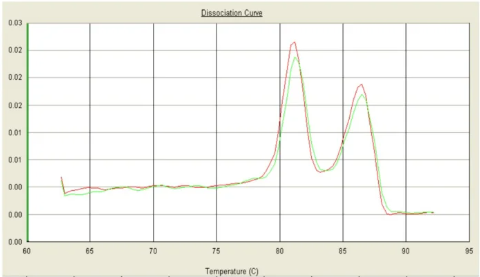
可能原因:
1. 引物特异性不好。
2. 空气中存在气溶胶污染。
解决方法:
1. 利用BLAST工具进行引物特异性检查,并根据检查结果重新设计引物。
2. 在严格的超净工作台中操作,采用专用的移液器分别加入引物,确保操作遵循无菌操作规范,减少交叉污染的风险。
九、其它非正常熔解曲线
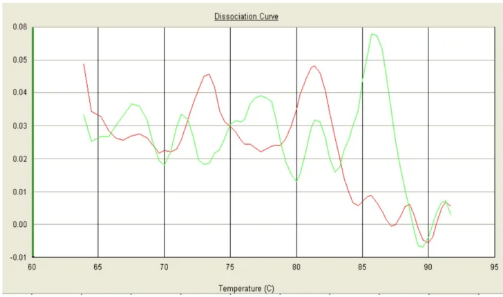
可能原因:
1. 体系受到污染,或试剂失去有效性。
2. 耗材与仪器之间不兼容,或检测通道选择不当。
3. 仪器长时间未进行必要的校准操作。
解决方法:
1. 在超净台内配制体系,避免污染。
2. 避免将试剂暴露于强光或高温环境中,以维持其稳定性,注意试剂限用时间,避免过期。
3. 执行阳性、阴性对照,选择与仪器完全匹配的耗材。
4. 定期仪器进行校准,确保其准确性和稳定性。
十、标准曲线线性关系不佳
可能原因:
1. 加样误差:加样过程中的操作误差使样本量不准确。
2. 标准品降解:标准品在存储或使用过程中发生降解。
3. 模板浓度高或有抑制物:模板的浓度过高或含有抑制PCR反应的成分,可能导致实验结果不准确。
4. 引物或探针不佳:引物或探针的设计不合理或质量不佳,可能无法有效进行PCR扩增或检测。
5. PCR酶的灵敏度可能因存储不当或过期而下降。
解决方法:
1. 严格控制标准品的加样体积,确保大于2ul。引物在使用前进行预混再分复孔使用。定期校正移液器,以减少误差。选择硅化枪头,以降低管壁对DNA的吸附。
2. 避免反复冻融,以减少降解。
3. 调整稀释精度,增加稀释梯度,以优化模板的浓度。
4. 重新设计引物和探针,确保其特异性。
更换灵敏度更高、线性关系更好的PCR试剂。同时:对-20℃冰箱进行校正,确保试剂的存储条件适宜。在使用酶时,应将其置于冰上,以保持其活性。
 文章推荐
文章推荐

业务咨询
业务咨询专线:133 7682 0615
Email:lxyjy@wie-biotech.com


