

代谢重编程是当前生命科学/基础医学研究的一大热点,近些年以“代谢重编程”为研究对象的高分文章也如“雨后春笋”般涌现。
继“糖代谢重编程”,这篇小编将带大家深入了解“脂肪酸代谢重编程”的相关研究思路。

【文章题目】:靶向MYC通过上调HILPDA诱导透明细胞肾细胞癌中脂滴聚集
【发表期刊】:美国国家科学院院刊
【影响因子】:IF=11.21
【发表日期】:2024.02
一、研究背景
代谢重编程在透明细胞肾细胞癌(ccRCC)的肿瘤发生过程中至关重要,其表现之一为脂滴(LD)在细胞器的积累。在缺乏VHL基因的ccRCC中MYC的抑制能以谷氨酰胺依赖的方式增加甘油三酯含量促进脂滴形成。此外,作者发现HILPDA是导致MYC驱动脂滴积聚的关键分子。最后通过对ccRCC及正常样本做对比,发现HILPDA可能是ccRCC的生物标志物。
二、研究思路
在本研究中,作者阐明ccRCC中靶向MYC通过上调HILPDA可以诱导脂质聚集。
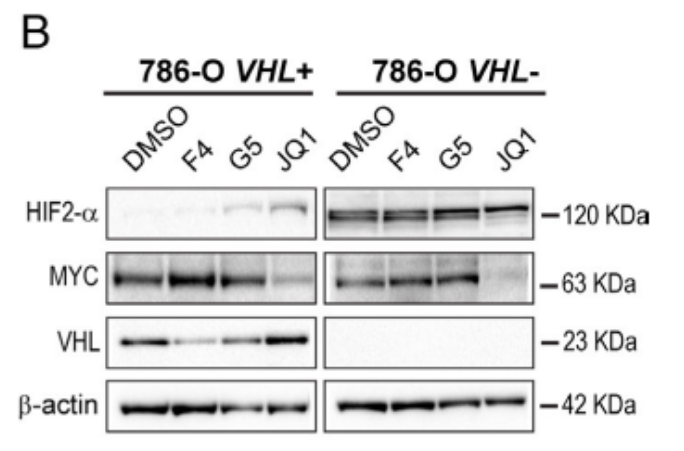
①首先验证了MYC抑制(多种抑制剂)可以促进脂质累积,包括VHL+/-细胞对MYC抑制的响应、HIF-1alpha的稳定性对MYC作用的影响以及谷氨酰胺及其酶对MYC作用的影响。
②然后通过脂质组学分析了MYC抑制处理VHL+/-细胞后的脂质代谢差异基因,以体内外样本证实差异基因HILPDA在MYC抑制中的作用。
③最后结合单细胞测序及数据库分析了差异基因HILPDA的价值。
三、研究结果
1. ccRCC中MYC抑制可使LD累积且需要HIF-1alpha参与
研究发现脂滴-苯并噻二唑1(LD-BTD1)染色脂质的染色方式并不影响细胞活力,也不影响MYC或HIF-alpha的表达。在图1A和图1B的实验中,作者采用3种抑制剂(F4,G5,JQ1)来抑制MYC的表达,结果显示MYC抑制之后脂滴累积增加。
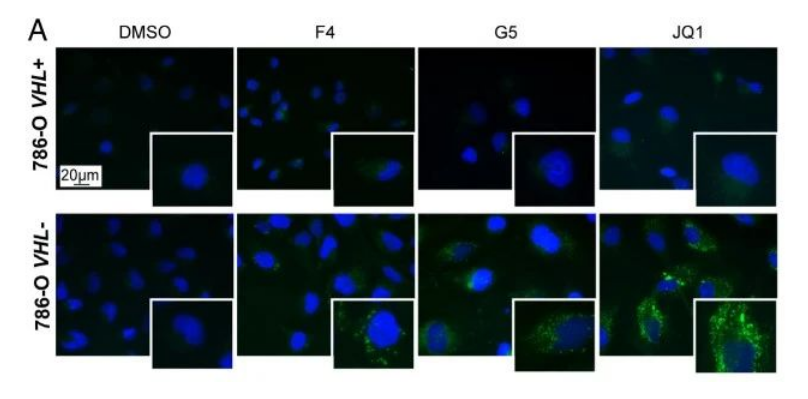
786-O代表人肾细胞癌
接着,作者诱导了Omomyc(MYC抑制)的表达,这是一种抑制MYC转录活性的显性负性肽,DOX多西环素处理可诱导HIF-1alpha,而图1C~E分别用DOX及缺氧两种方式诱导HIF-1alpha。图1E显示,在VHL+细胞中通过缺氧诱导了HIF之后,MYC抑制同样上调了脂质积聚。最终,研究结果表明,只有在VHL缺失的ccRCC细胞中,LD才会在药理或遗传性MYC抑制后积累。
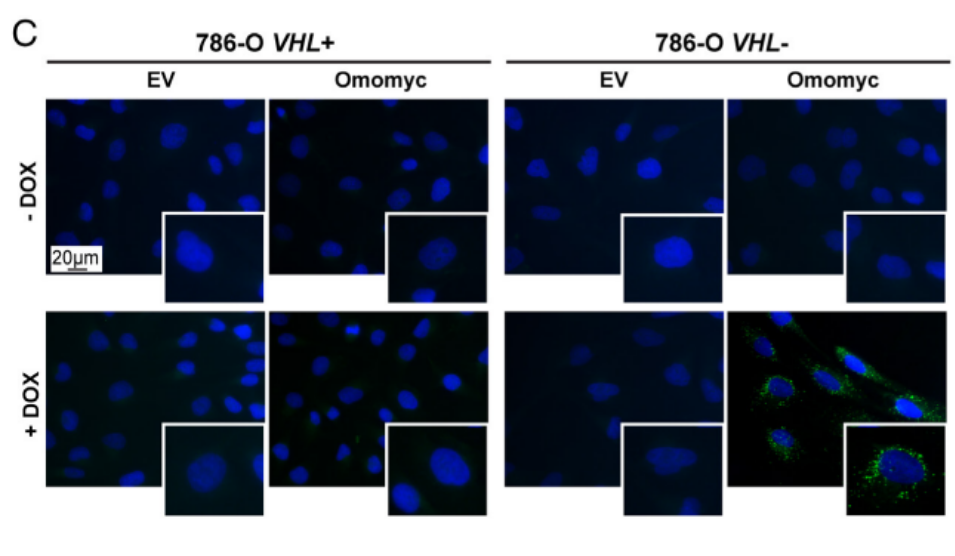
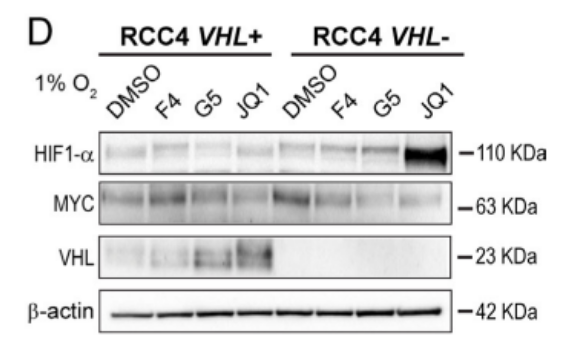
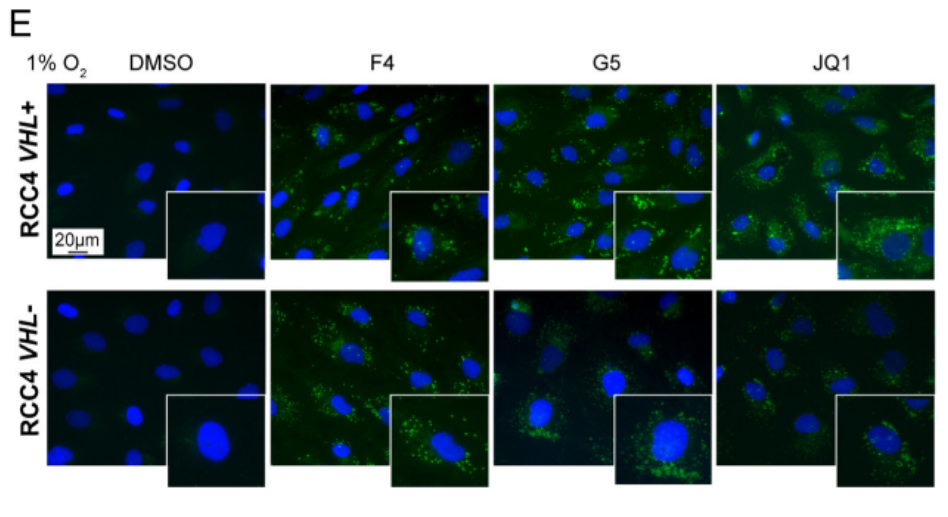
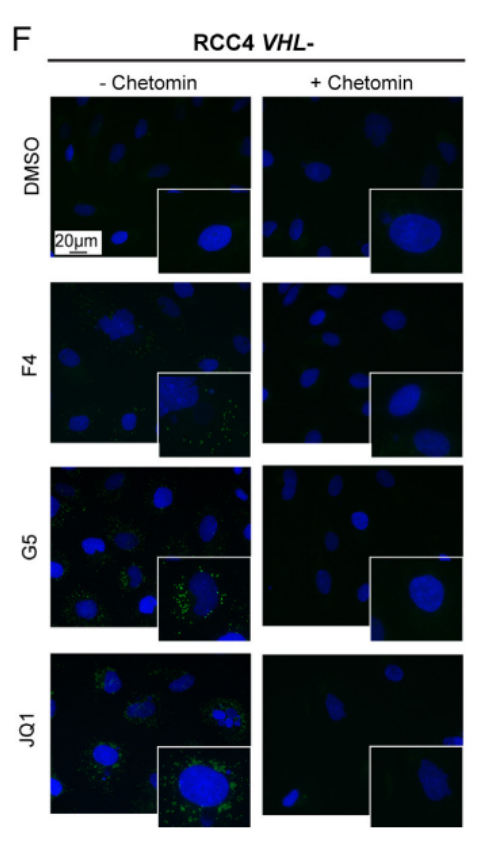
图1F显示,使用Chetomin抑制HIF与其转录共激活因子p300和cAMP反应元件结合(CREB)的结合蛋白(CBP)的相互作用,在RCC4 VHL-细胞中联合MYCis时可以抑制脂肪的合成。这说明在MYCis存在时引入HIFs会导致脂肪积累无论在VHL+和VHL-的ccRCC细胞中,这一过程会被HIF抑制所阻止。
2. 谷氨酰胺水平低或谷氨酰胺酶抑制可阻碍LD的形成
因为储存在LD中的脂肪酸可以从细胞外环境中获得,也可以由葡萄糖或谷氨酰胺合成,因此作者进一步探究用于LD形成的燃料。图2A、2B显示,通过降低培养基中谷氨酰胺水平(Gln)及抑制谷氨酰胺酶抑制剂,可抑制MYC抑制诱导的LD积聚。
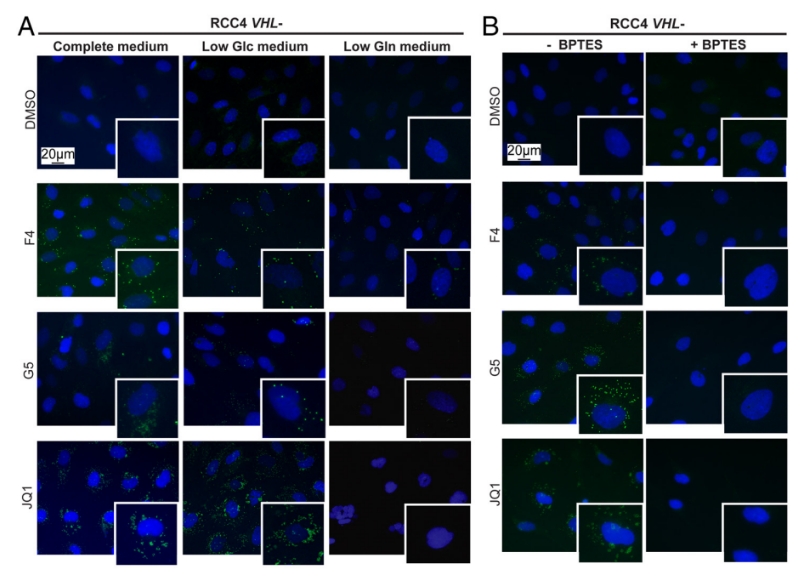
图2C同样显示,通过抑制谷氨酰胺酶抑制剂,可抑制Omomyc诱导的脂质积聚。综合以上数据,可见谷氨酰胺是MYC抑制后脂肪酸合成和存活所必需的。
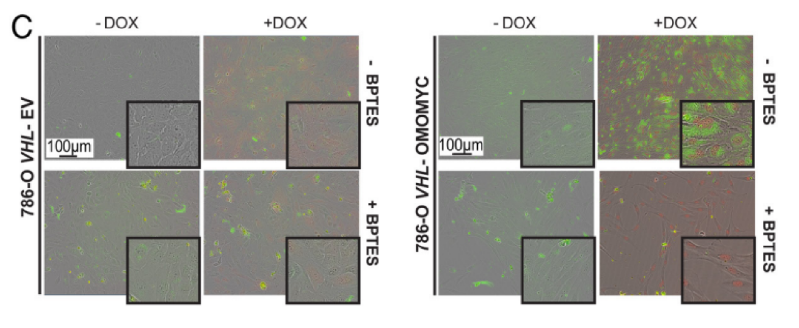
3. MYC抑制表现在VHL+细胞和VHL−细胞中不同脂质物种的积累
为了研究谷氨酰胺衍生的碳是否直接用于脂肪酸合成,作者在RCC4 VHL+和VHL−细胞中进行了稳定同位素示踪实验。JQ1处理略微降低了RCC4 VHL−细胞中葡萄糖和谷氨酰胺对乙酰辅酶a的贡献。然而,在MYC抑制后,谷氨酰胺衍生的乙酰辅酶a没有发生变化,这表明JQ1降低了葡萄糖对乙酰辅酶a的贡献。图3A显示,在VHL−细胞中,谷氨酰胺衍生的碳用于棕榈酸盐合成的程度远远大于VHL+细胞。
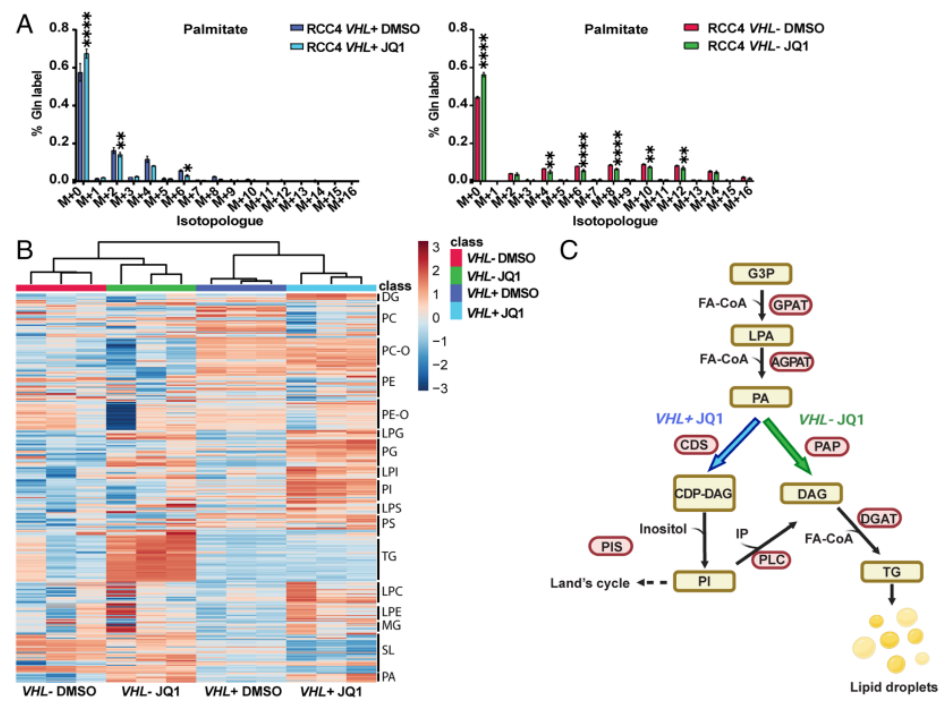
然后,作者通过脂质组学分析了VHL+-JQ1处理后的差异代谢产物,图3B和3C结果显示,用JQ1处理的RCC4 VHF-细胞中的甘油三酯(TGs)更丰富,而RCC4 VHF+细胞中溶血磷脂及甘油磷脂更为丰富。综上所述,作者发现RCC4 VHL−细胞使用谷氨酰胺产生乙酰辅酶a用于脂质合成,而RCC4 VHL+细胞使用葡萄糖。重要的是,MYC抑制仅在VHL -细胞中诱导TG积累,这解释了VHL+和VHL-ccRCC细胞之间LD丰度的差异。
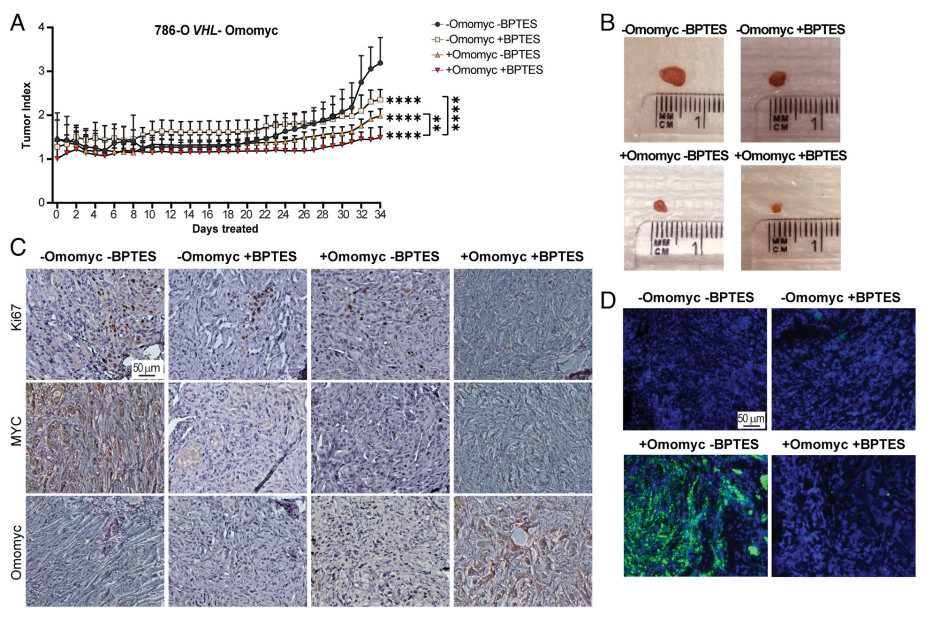
4. 联合抑制MYC信号通路和谷氨酰胺代谢可抑制肿瘤的生长
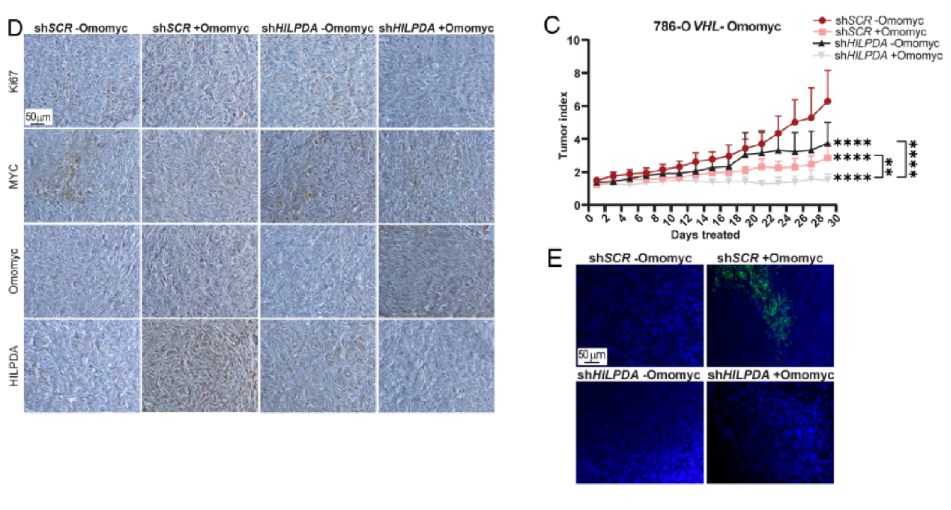
接着,作者为了分析MYC和GLS联合抑制LD积累和体内肿瘤生长的相关性,做了荷瘤实验。我们可以从图4A~D中发现,MYC抑制并且HIF未抑制的情况下瘤体内脂质积聚更多。而MYC抑制合并HIF抑制的瘤体更小,这是因为MYC和HIF均有促进增殖的作用。最终,作者认为Omomyc抑制MYC也诱导了VHL缺陷的ccRCC细胞体内LD的形成,而Omomyc的表达和谷氨酰胺代谢的抑制阻止了它们的形成并进一步减轻了肿瘤负荷。
5. 脂滴相关蛋白在VHL-的细胞中表达上调
然后,作者进行了大量RNA测序(RNA-seq)分析,以确定在HIF表达的细胞中,MYC抑制后导致LD积累的分子机制。图5A显示,与对照相比,在JQ1处理后的RCC4 VHL-细胞中有515个基因下调,308个基因上调,而RCC4 VHL+细胞中有629个基因下调,215个基因上调。此外,图5B~C显示,差异基因还是较多富集在脂质代谢中,且低氧诱导的脂滴相关(HILPDA)基因是RCC4 VHL-细胞中抑制MYC时最显著上调的基因之一。
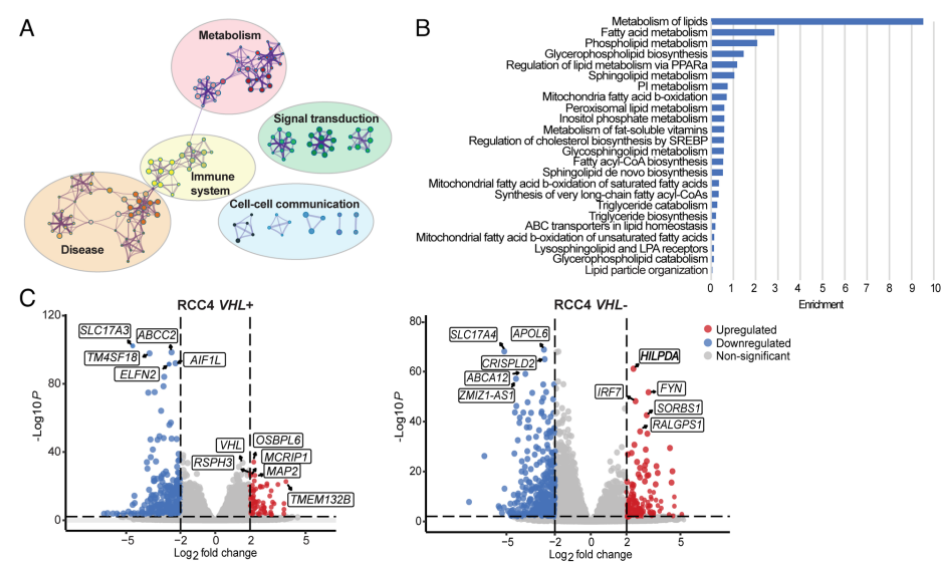
接下来,作者进一步对体内外的样本进行检验。图5C~E显示,HILPDA在MYC抑制时表达下调。综合以上研究结果,可知在HIF激活和MYC抑制的联合作用下,HILPDA表达上调,这与VHL−细胞中LD的积累有关。
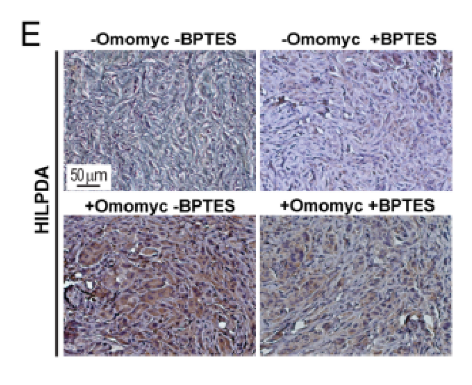
6. 选择性表达表明HILPDA是ccRCC中的一种新的易治疗点
为了研究在MYC抑制后诱导LD积累是否需要HILPDA表达,在图5F~G中作者使用siRNA下调其表达,并观察到在RCC4 VHL -细胞中LD的形成量降低了。此外,图5H显示,敲低HILPDA后,细胞中的HILPDA水平下调了95%,并且LD的积累也降低了。图6A、6B则显示,过表达HILPDA能促进VHL-细胞中LD积聚。
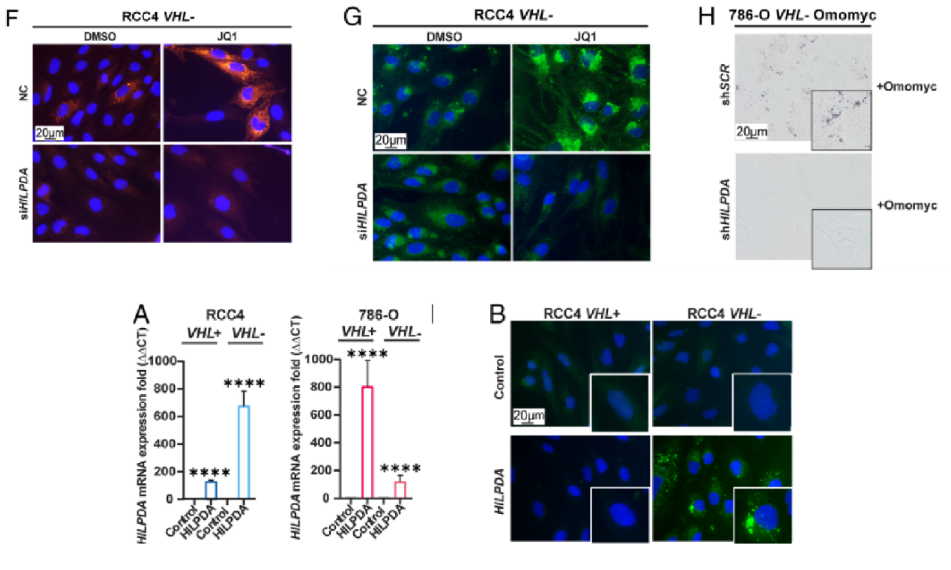
为了研究HILPDA下调对体内肿瘤生长和LD积累的影响,作者又使用786-O VHL−Omomyc shHILPDA和shSCR细胞进行了异种移植实验。图6C~E结果,沉默HILPDA能抑制MYC抑制导致的LD累积,但shHILPDA能进一步下调MYC抑制的瘤体体积。
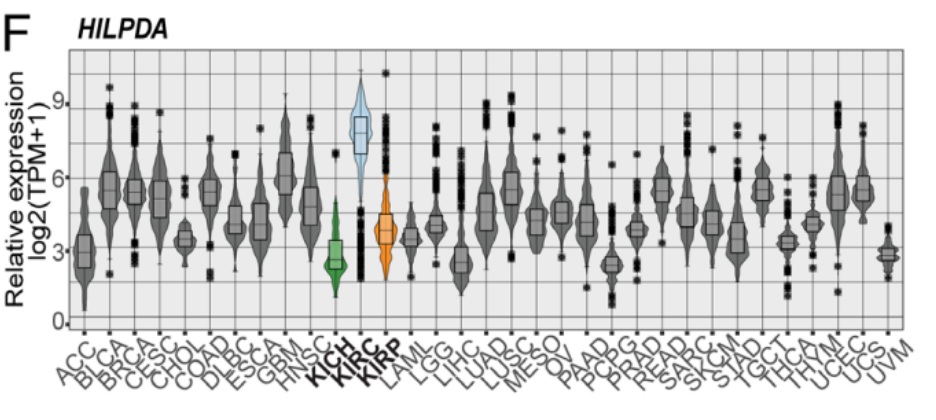
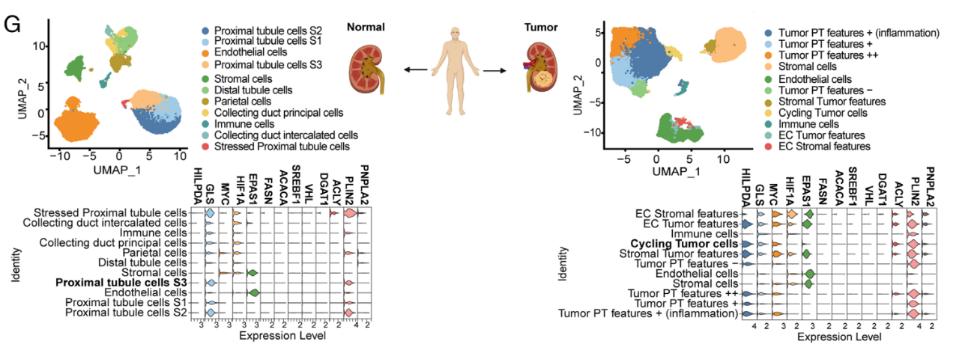
为了调查研究结果可以转化为临床样本的可能性,作者使用了来自癌症基因组图谱(TCGA)的公开数据,图6F的数据分析结果显示,HILPDA在ccRCC(KIRC)中的选择性高于其他所有肿瘤,包括其他两种:嫌色(KICH)绿和乳头状(KIRP)橙。
最后,作者分析了来自患者正常肾脏和肿瘤组织的单细胞RNA-seq(scRNA-seq)。图6G数据结果显示,尽管循环肿瘤细胞以及来自肿瘤间质的其他细胞显示出高水平的HILPDA,但来自健康肾组织的细胞中没有表达该基因。此外,MYC水平仅在肿瘤细胞中观察到,特别是在具有增殖特征的肿瘤细胞中。与TCGA数据库的数据类似,与正常组织相比,肿瘤中GLS、ACLY和PLIN2升高,而VHL和编码脂肪酸合成途径中其他酶的基因未被检测到。最后,编码HIF2-α的EPAS1主要在肿瘤中表达,肿瘤细胞与正常组织中HIF-1A的表达无明显差异。综合以上,作者确定HILPDA是ccRCC的一种特异性生物标志物,因为它在癌症和肿瘤相关的间质细胞中都有表达,但在正常肾脏中不表达。
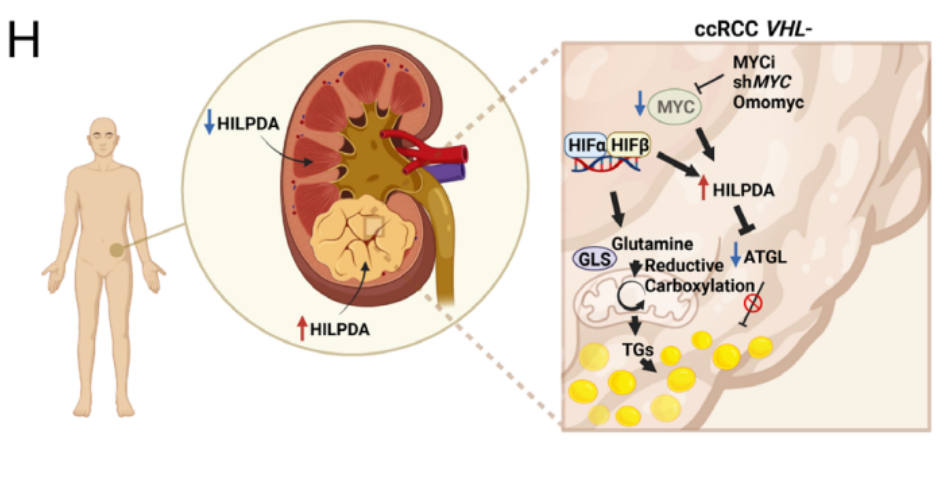
此外,正常肾组织中HILPDA表达的缺失为靶向ccRCC肿瘤而不破坏周围健康组织提供了可能性。总的来说,该研究确定了导致LD在ccRCC中强劲积累的分子机制,这是由于HIF同时表达和MYC抑制涉及HILPDA时TG含量的增加。随着LD在癌症进展和生存中的作用越来越重要,了解LD积累的过程,以及确定HILPDA等选择性靶点,可以为ccRCC的患者精准医疗策略奠定基础。
 文章推荐
文章推荐

业务咨询
业务咨询专线:133 7682 0615
Email:lxyjy@wie-biotech.com


