

慢阻肺(COPD)是慢性阻塞性肺疾病的简称,一种以持续呼吸道症状和气流受限为特征的慢性气道和肺疾病,也是一种可以预防的慢性气道炎症性疾病,虽然是气道疾病,但对全身各系统的影响也是不容忽视的。那么关于慢阻肺有什么课题设计思路呢?今天我们一起来探讨一下。
下面,我们将分享一篇发表于中科院1区期刊Nature Communications,影响因子为14.7的高分文献,名为《香烟烟雾诱导的上皮细胞铁死亡在慢性阻塞性肺疾病发病中的作用》,希望能给大家带来不一样的灵感。

【文章题目】:香烟烟雾诱导的上皮细胞铁死亡在慢性阻塞性肺疾病发病中的作用
【发表期刊】:Nature Communications
【影响因子】:IF=14.7
【发表日期】:2019.07
一、研究背景
①铁死亡被认为是一种铁依赖性调控细胞死亡(RCD),其特征是铁介导的Fenton反应中产生的活性氧(ROS)引起质膜磷脂过氧化。
②在肾脏和脑损伤模型中已经证实了铁死亡在疾病发病机制中的作用。
③慢性香烟烟雾(CS)暴露引起的异常炎症过程已被认为是COPD发病机制的一部分。
④CS已被证明不仅能诱导细胞凋亡,还能诱导坏死和随后从气道上皮细胞释放DAMPs,这与气道炎症有因果关系。
⑤铁自噬依赖于选择性货物受体NCOA4,它将铁蛋白运输到自噬体并促进铁死亡。
二、技术路线
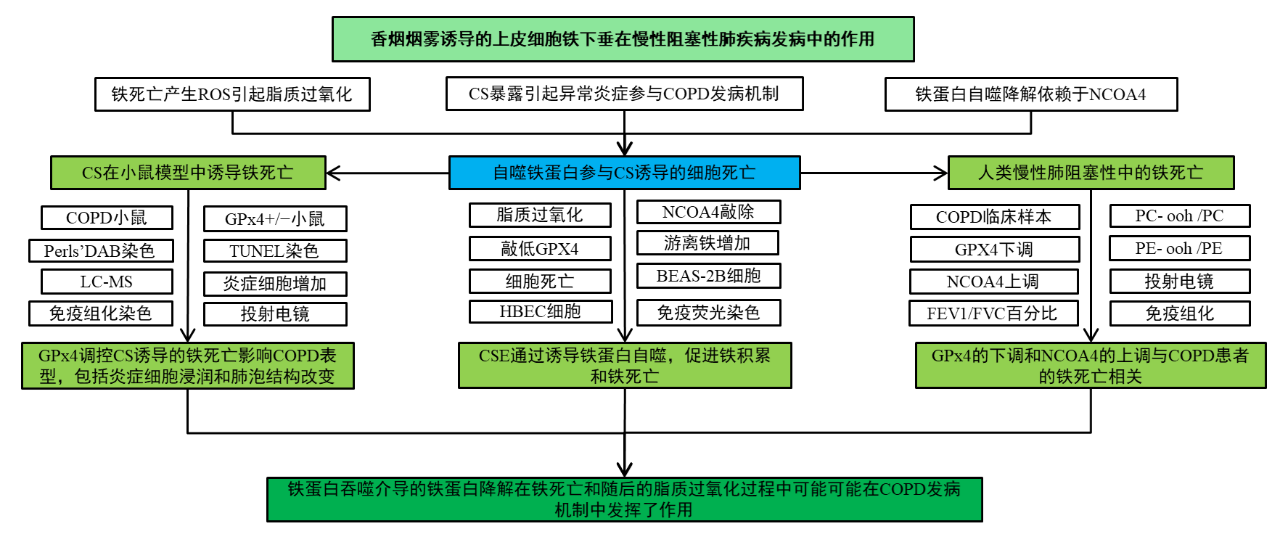
三、研究结果
1.香烟烟雾提取物(CSE)诱导人支气管上皮细胞(HBECs)铁死亡
为了阐明铁死亡在CSE介导的细胞死亡中的作用,作者在HBECS中处理24h后,通过C11BODIPY染色评估CS诱导的脂质过氧化。图1a~f的C11BODIPY染色显示,CSE暴露后有明显的脂质过氧化反应,而去铁胺(DFO)明显消除了CSE诱导的脂质过氧化,并且抑制CSE诱导的细胞死亡。我们可以从图1c~d的实验结果看到,Fer-1也能显著减弱CSE诱导的脂质过氧化和细胞死亡。然而,如图1d所示,坏死性凋亡抑制剂Nec1和pan-caspase抑制剂zVAD-FMK对CSE诱导的细胞死亡的影响可以忽略不计,这进一步支持了铁死亡参与CSE诱导的细胞死亡。
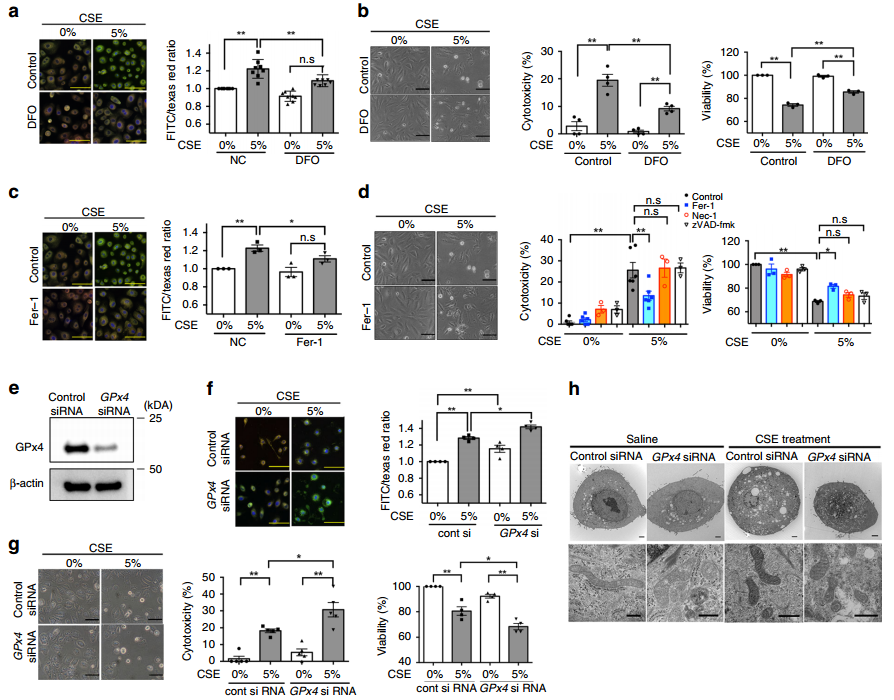
接下来,我们可以从图1f和1g看到,敲低GPx4显著增强了CSE诱导的HBECs细胞的脂质过氧化和细胞死亡。在透射电子显微镜(TEM)下,CSE处理的HBECs线粒体致密和萎缩非常明显,是典型的铁死亡形态特征。并且,我们可以从图1h看到,CSE诱导的线粒体膜密度增加在GPx4敲低中更为突出。作者推论,在此实验条件下铁死亡是CSE诱导细胞死亡的主要原因。
2.铁自噬参与CS诱导的细胞死亡
为了阐明游离铁积累与COPD发病机制中铁死亡诱导相关的分子机制,作者重点关注了NCOA4介导的CSE处理支气管上皮细胞的铁自噬。我们可以从图2a~b结果观察到,CSE处理瞬间增加了铁蛋白的表达,并在6h时转变为时间依赖性的衰减,同时BEAS-2B中NCOA4的表达上调。钙黄素-AM测定证实,5%CSE处理后,如图2c所示,游离铁浓度增加,NCOA4敲除降低了游离铁浓度。图2d显示,NCOA4的敲除增强了CSE处理的细胞内铁蛋白积累,进一步表明CSE暴露期间铁自噬参与铁蛋白降解。
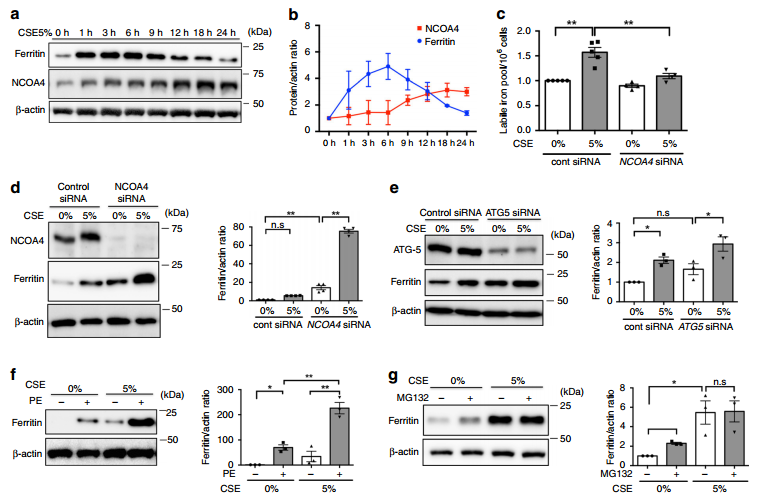
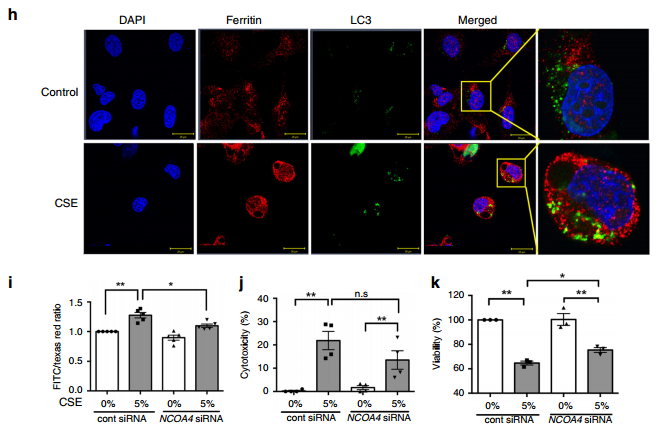
我们还可以从图2e~f观察到,敲低ATG5和使用Pepstatin A和E64抑制自噬后,铁蛋白积累也增强了。图2g显示,用蛋白酶体抑制剂MG132处理后,没有观察到铁蛋白显著增加,这表明在CSE暴露期间,蛋白酶体不参与铁蛋白降解。图2h的BEAS-2B免疫荧光染色显示,CSE处理增强了铁蛋白和LC3-GFP的表达,并检测到铁蛋白与LC3-GFP共定位。此外,我们可以从图2i~k发现,BEAS-2B中NCOA4的下调减少了CSE诱导的脂质过氧化和细胞死亡,表明通过NCOA4介导的铁自噬降解铁蛋白是CS暴露期间支气管细胞死亡中的铁死亡的原因。综合上述表明,CSE通过诱导铁自噬,促进铁积累和铁死亡,其中NCOA4和GPx4的相互作用在调控铁死亡中起关键作用。
3.CS在小鼠模型中诱导铁死亡
接着,作者评估了CS是否增加了COPD实验小鼠模型中的细胞内不稳定铁。图3a结果显示,Perls的DAB染色显示CS暴露小鼠的支气管上皮细胞具有更高水平的非血红素铁。并且,图3b~d显示,CS暴露小鼠气道和全肺匀浆中的游离铁水平显著升高;同体外实验一致,CS暴露小鼠的肺匀浆中铁蛋白的表达明显低于RA暴露的肺匀浆,并且CS暴露小鼠肺匀浆中NCOA4表达水平显著升高。
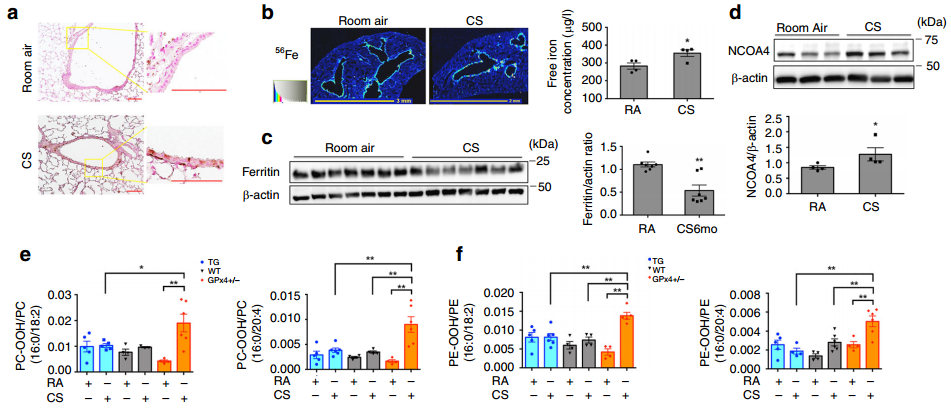
此外,图3e~f的LC-MS/MS显示,CS暴露增加GPx4+/−小鼠PC-OOH/PC比率和PE-OOH/PE比率。图3g的免疫染色显示,CS暴露使WT小鼠和GPx4+/−小鼠的4-HNE染色增强,而GPx4 TG小鼠的4-HNE染色在气道上皮细胞和肺泡上皮细胞中均降低。我们还可以从图3h观察到,TUNEL阳性细胞在CS暴露的WT小鼠肺中略有但显著增加,GPx4+/−小鼠肺中TUNEL阳性细胞相对于WT显著增强,GPx4 TG小鼠肺中TUNEL阳性细胞相对于WT显著减少。CS暴露后,如图3i~j所示,肺均质液中Cleaved caspase-3蛋白水平也略有升高,但在GPx4+/-和GPx4 TG小鼠中未检测到改变,表明CS暴露的GPx4+/-小鼠中细胞死亡的显著增加主要归因于铁死亡。以上实验结果表明CS诱导的铁死亡涉及GPx4调控的脂质过氧化和NCOA4介导的铁蛋白自噬。
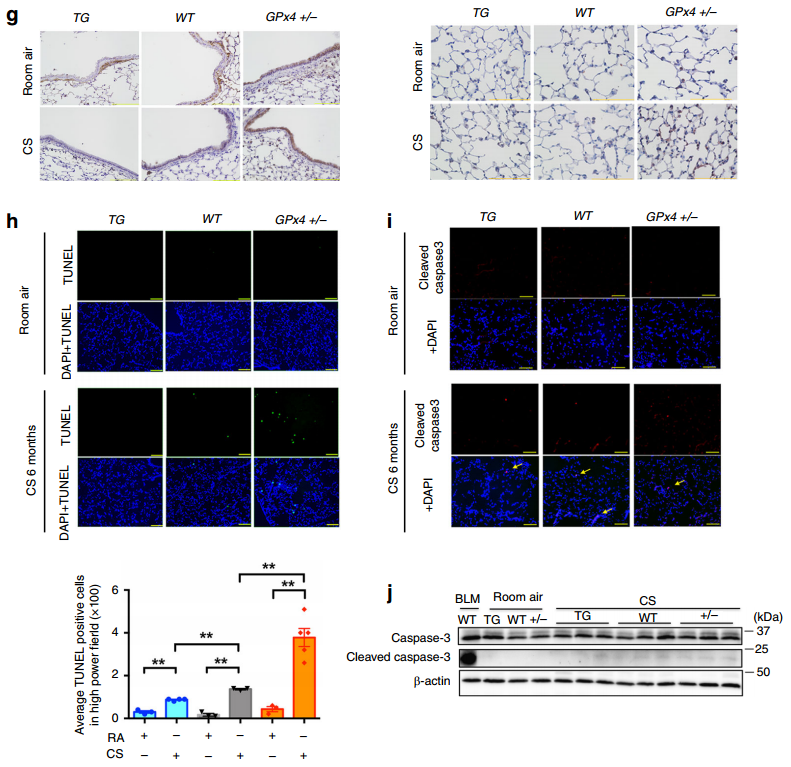
4.GPx4调节吸烟小鼠模型中的COPD表型
然后,作者统计了各组小鼠在CS暴露后肺部支气管肺泡灌洗液(BALF)中获得的炎症细胞。GPx4+/−小鼠的BALF中总细胞、巨噬细胞和淋巴细胞数进一步增加。我们可以从图4a发现,相比之下,GPx4 TG小鼠的BALF中未观察到细胞计数的显著增加。图4b~d显示,WT小鼠肺均质液中的DAMPs,包括IL-33和IL-1α显著增加,GPx4+/−小鼠的DAMPs进一步增强,而GPx4 TG小鼠的BALF中HMGB-1显著减少。此外,促炎细胞因子TNF-α的表达升高。但是图4e结果显示,TNF-α上调在GPx4 TG小鼠中被抑制。总的来说,CS暴露诱导铁死亡,并伴有肺上皮细胞释放DAMPs和促炎细胞因子。
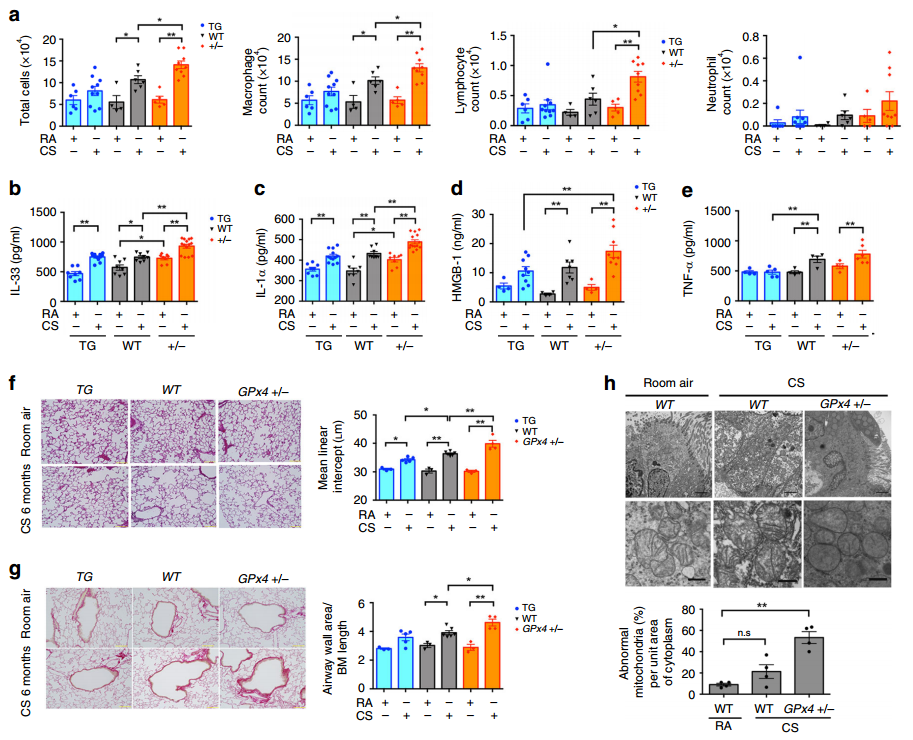
此外,图4f~g显示,通过肺泡平均线性截距(Lm)测量,CS暴露导致WT小鼠肺空域明显增大、气道壁增厚增加,GPx4+/−小鼠肺空域扩大增强、气道壁增厚更明显,而GPx4 TG小鼠肺空域扩大明显减弱、气道壁增厚未见改变。接着从图4h观察到,在CS暴露的WT小鼠中,线粒体的积累随着膜密度的增加而增加,而在GPx4+/−小鼠中,线粒体的积累与膜密度增加同时伴有嵴断裂,这是一个突出的特征。最终表明GPx4调控CS诱导的铁死亡影响COPD表型,包括炎症细胞浸润和肺泡结构改变。
5.人类慢性阻塞性肺疾病中铁死亡的参与
为了阐明GPx4在COPD发病机制中的潜在参与,作者收集了不吸烟者、吸烟者和COPD患者的人肺支气管上皮细胞,并检测了GPx4的表达水平。图5a的WB显示,来自COPD肺的HBECS中GPx4的表达明显降低;此外,GPx4表达水平与FEV1/FVC百分比呈正相关。图5b的肺组织免疫组化也显示,COPD患者支气管上皮细胞中GPx4的表达低于非COPD患者。
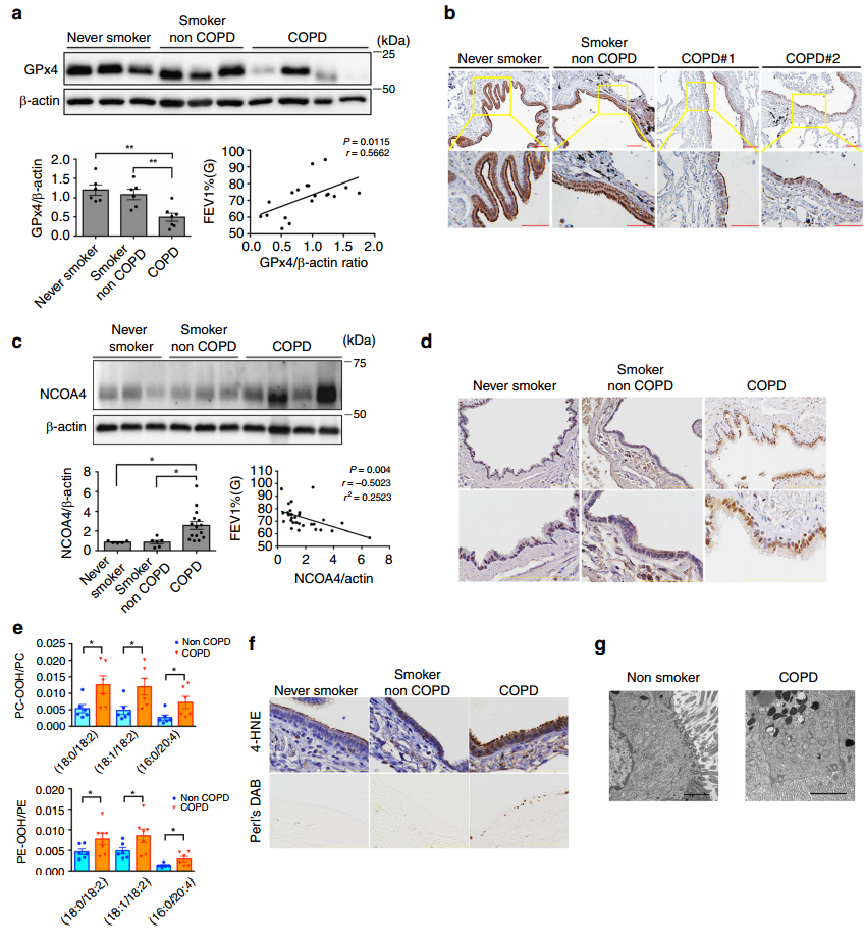
接下来我们可以从图5c发现,COPD患者肺匀浆中NCOA4表达水平显著升高。而图5c显示,NCOA4蛋白水平与FEV1/FVC百分比呈负相关。图5d~f显示,支气管上皮细胞中NCOA4的表达增强,特别是在COPD患者中;此外,COPD肺中检测到明显的4-HNE阳性染色,主要在支气管上皮细胞的细胞质中,同时perls'DAB染色增强,证明了肺上皮细胞中游离铁的增加。图5e则显示,COPD肺匀浆中PC-OOH/PC比率和PE-OOH/PE显著高于非COPD肺匀浆。并且在COPD肺的气道上皮细胞中,如图5g所示,随着膜密度增加,小尺寸线粒体的积累很明显,但在非吸烟者肺的气道上皮细胞中几乎没有检测到。以上实验结果表明GPx4的下调和NCOA4的上调与COPD患者的铁死亡相关,暗示铁死亡可能参与COPD的病理过程。
四、研究小结
该研究证明了铁死亡参与COPD的发病机制。作者通过体内和体外模型发现,在CS暴露期间,不稳定的铁积累和脂质过氧化增强,伴随非凋亡细胞死亡,这是受GPx4活性的负调控。除GPx4敲除外,用DFO和Fer-1治疗可阐明铁死亡在CS处理的肺上皮细胞中的作用。NCOA4介导铁自噬在CS治疗后的铁蛋白降解过程中启动。CS暴露模型,使用GPx4缺陷和过表达小鼠,阐明了GPx4调节的细胞死亡在COPD中的关键作用。这些发现支持了CS诱导的铁死亡在COPD发病机制中的作用。
五、国自然中标情况

 文章推荐
文章推荐

业务咨询
业务咨询专线:133 7682 0615
Email:lxyjy@wie-biotech.com


